
El sistema de reparación de apareamiento incorrecto de ADN se conoce con las siglas en inglés de MMR (mismatch repair). Este sistema detecta y corrige errores que ocurren durante la replicación del ADN, como apareamientos incorrectos de bases o las inserciones y deleciones. Este complejo sistema está compuesto por los genes que codifican las proteínas MLH1, MSH2, MSH6 y PMS1 y el gen MSI.
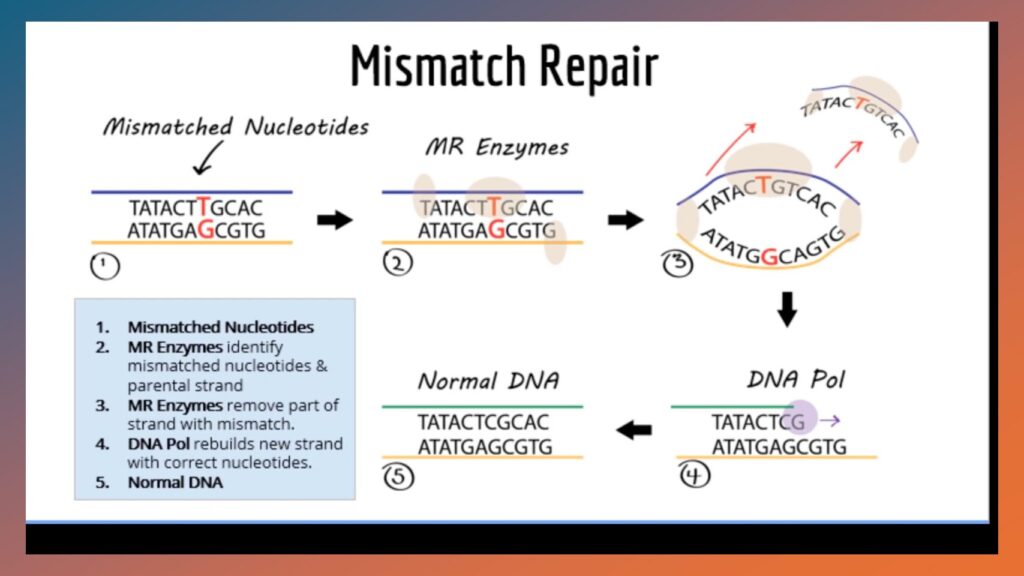
Cuando el ADN se está duplicando, las enzimas MMR detectan los nucleotidos mal emparejados, estas enzimas remueven el segmento de hebra del ADN con el error, posteriormente la ADN polimerasa reconstruye el segmento de hebra faltante y restituye el ADN a la normalidad.
La inestabilidad microsatelital (MSI) es una condición genética caracterizada por cambios en la longitud de secuencias repetitivas de ADN, llamadas microsatélites, esto es debido a errores en la replicación del ADN que no son corregidos. Los microsatélites son secuencias cortas de ADN que se repiten varias veces y están presentes en todo el genoma. Durante la replicación del ADN, el sistema MMR corrige los errores de apareamiento que ocurren en estas secuencias. Cuando el sistema de reparación MMR está defectuoso (debido a mutaciones en genes como MLH1, MSH2, MSH6 o PMS2), los errores de replicación en los microsatélites se acumulan, resultando en variaciones de longitud de estas secuencias en las células tumorales comparadas con las células normales.
El MSH2 y MSH6 se asocian para reconocer las bases mal alineadas y unirse al sitio del error, luego el MLH1 y PMS2 intervienen para coordinar la escisión y resíntesis de la región defectuosa. Esta maquinaria elimina los nucleótidos erróneos y permite la reincorporación de las bases correctas. De esta manera se preserva la integridad genómica al asegurar la fidelidad en la alineación de las bases.
¿Cómo se estudian estos genes?
La evaluación de estos genes puede hacerse con Inmunohistoquímica y pruebas genómicas.
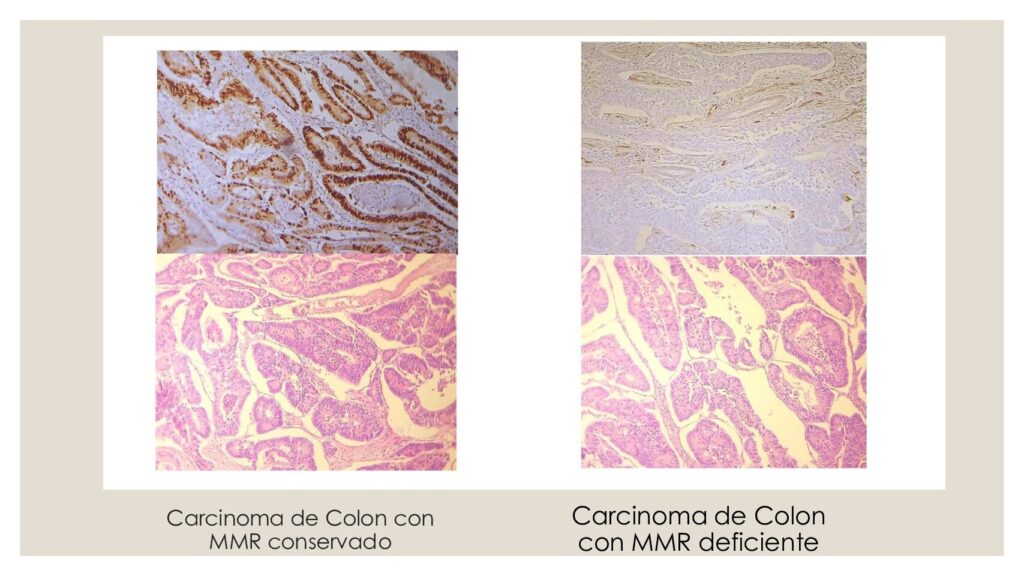
Está validado hacer el estudio de estas proteínas por Inmunohistoquímica, se busca la pérdida de la expresión de estas proteínas en las células neoplásicas, el algoritmo de interpretación es como sigue:
1. Pérdida de MLH1 y PMS2:
– Indica deficiencia en MLH1.
– Interpretación clínica: La pérdida de MLH1 generalmente implica que el tumor es deficiente en reparación de ADN por una alteración en MLH1. Esta pérdida puede estar relacionada con metilación del promotor de MLH1 o con una mutación en el gen MLH1.
– Próximo paso: Si se sospecha síndrome de Lynch, se puede realizar una prueba para detectar la metilación del promotor de MLH1 y/o análisis de inestabilidad de microsatélites.
2. Pérdida de MSH2 y MSH6:
– Indica deficiencia en MSH2.
– Interpretación clínica: La pérdida de MSH2 implica que el tumor es deficiente en MMR debido a una alteración en el gen MSH2. Esto puede estar asociado a una mutación en MSH2, típicamente relacionada con el síndrome de Lynch.
– Próximo paso: Evaluar antecedentes familiares y considerar pruebas genéticas para confirmar el síndrome de Lynch.
3. Pérdida de MSH6 únicamente:
– Indica deficiencia en MSH6.
– Interpretación clínica: La pérdida aislada de MSH6 es menos común, pero puede estar asociada con mutaciones en el gen MSH6. Esto también puede estar relacionado con el síndrome de Lynch, aunque es menos frecuente.
– Próximo paso: Evaluar antecedentes familiares y considerar pruebas genéticas.
4. Pérdida de PMS2 únicamente:
– Indica deficiencia en PMS2.
– Interpretación clínica: La pérdida aislada de PMS2 sugiere una alteración en el gen PMS2. Esto también puede estar relacionado con el síndrome de Lynch.
– Próximo paso: Evaluar antecedentes familiares y considerar pruebas genéticas.
5. Tinción positiva para todos los marcadores (MLH1, MSH2, MSH6, PMS2):
– Interpretación clínica: La expresión normal de todos los genes MMR sugiere que el sistema de reparación de errores de apareamiento de ADN es funcional.
– Próximo paso: No se requieren pruebas adicionales de MMR por inmunohistoquímica; considerar otros métodos diagnósticos si el contexto clínico lo requiere.
La detección de la reparación defectuosa de errores de apareamiento en los carcinomas colorrectales es importante para el diagnóstico del síndrome de Lynch (síndrome de cáncer colorrectal hereditario no polipósico-CCRNP), que representa aproximadamente el 2% al 3% de todos los carcinomas colorrectales y tiene implicaciones clínicas en el tratamiento del paciente afectado y de sus familiares. Adicionalmente hoy en día está indicada descartar el Síndrome de Lynch en pacientes con diagnóstico de Adenocarcinoma endometrioide.
El diagnóstico de inestabilidad de microsatélites se hace por pruebas genómicas que detectan mutaciones en MSI, los pacientes con un fenotipo MSI-H (Alta inestabilidad de microsatélites) tienen una mutación en los genes MMR o el gen EPCAM (TACSTD1). Tras una adecuada asesoría genética, los pacientes pueden considerar pruebas para identificar la anomalía hereditaria causante. Un fenotipo MSI-H se observa más frecuentemente en el cáncer colorrectal esporádico (alrededor del 15% de los casos) debido a anormalidades somáticas, generalmente hipermetilación del promotor del gen MLH1.
HIPERMETILACIÓN DE MSI (SILENCIAMIENTO EPIGENETICO):
La reparación defectuosa de errores de apareamiento en el cáncer colorrectal esporádico suele deberse a la inactivación del promotor del gen MLH1 por hipermetilación (silenciamiento epigenético). La mutación V600E del gen BRAF puede estar presente en hasta el 70% de los tumores con hipermetilación del promotor de MLH1. La prueba directa de hipermetilación del promotor de MLH1 y/o el uso del análisis de mutación BRAF V600E antes de las pruebas genéticas germinales en pacientes con tumores MSI-H y pérdida de MLH1 por inmunohistoquímica pueden ser un método mas económico para identificar pacientes con tumores esporádicos para quienes no se indica realizar pruebas adicionales.
En conclusión, la evaluación de la inestabilidad microsatelital y la expresión de las proteínas MMR es clave para diagnosticar el síndrome de Lynch y diferenciarlo de los casos esporádicos de cáncer colorrectal, esto permite orientar el tratamiento de forma más precisa y brindar asesoramiento genético adecuado a los pacientes y sus familias. Además, la identificación de alteraciones epigenéticas (como la hipermetilación del promotor de MLH1) o mutaciones específicas (como BRAF V600E) ayuda a distinguir entre tumores hereditarios y esporádicos, mejorando así la toma de decisiones clínicas y el beneficio de tratamiento de precisión o personalizado.
BIBLIOGRAFIA:
1- Template for Reporting Results of Biomarker Testing of Specimens From Patients With Carcinoma of the Colon and Rectum. ColoRectal.Bmk_1.3.0.0.REL_CAPCP.
2- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261-268.
3- Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology Provisional Clinical Opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27(12):2091-2096.
4- Domingo E, Niessen RC, Oliveira C, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24(24):3995-3998.